 |
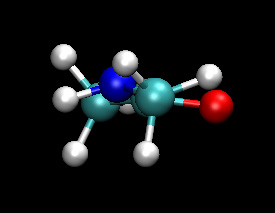 |
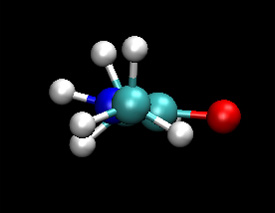
| Classical (MM) | QM |
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2004
TUTORIAL 6 - AMBER 9 VERSION - SECTION 4
A Coupled Potential QM/MM Simulation
By Ross Walker
PLEASE NOTE THIS TUTORIAL IS FOR AMBER 9 ONLY.
4) Comparing the results
We will now qualitatively compare the results of the two simulations. First of all lets take a brief look at the two output files:
|
Classical |
NSTEP = 0 TIME(PS) = 0.000 TEMP(K) = 455.28 PRESS = 0.0 Etot = -9086.7258 EKtot = 4144.0991 EPtot = -13230.8249 BOND = 0.0650 ANGLE = 0.1616 DIHED = 2.5068 1-4 NB = 1.1157 1-4 EEL = -19.4429 VDWAALS = 930.8446 EELEC = -14146.0758 EHBOND = 0.0000 RESTRAINT = 0.0000 Ewald error estimate: 0.1330E-03 ------------------------------------------------------------------------------ NSTEP = 1 TIME(PS) = 0.002 TEMP(K) = 346.22 PRESS = 0.0 Etot = -10079.4435 EKtot = 3151.3814 EPtot = -13230.8249 BOND = 0.0650 ANGLE = 0.1616 DIHED = 2.5068 1-4 NB = 1.1157 1-4 EEL = -19.4429 VDWAALS = 930.8446 EELEC = -14146.0758 EHBOND = 0.0000 RESTRAINT = 0.0000 Ewald error estimate: 0.1330E-03 ------------------------------------------------------------------------------ |
|
QMMM |
NSTEP = 0 TIME(PS) = 0.000 TEMP(K) = 455.28 PRESS = 0.0 Etot = -9041.3222 EKtot = 4144.0991 EPtot = -13185.4213 BOND = 0.0000 ANGLE = 0.0000 DIHED = 0.0000 1-4 NB = 0.0000 1-4 EEL = 0.0000 VDWAALS = 881.6951 EELEC = -14012.9122 EHBOND = 0.0000 RESTRAINT = 0.0000 PM3ESCF= -54.2042 Ewald error estimate: 0.5641E-01 ------------------------------------------------------------------------------ NSTEP = 1 TIME(PS) = 0.002 TEMP(K) = 346.23 PRESS = 0.0 Etot = -10033.9525 EKtot = 3151.4687 EPtot = -13185.4213 BOND = 0.0000 ANGLE = 0.0000 DIHED = 0.0000 1-4 NB = 0.0000 1-4 EEL = 0.0000 VDWAALS = 881.6951 EELEC = -14012.9122 EHBOND = 0.0000 RESTRAINT = 0.0000 PM3ESCF= -54.2042 Ewald error estimate: 0.5641E-01 ------------------------------------------------------------------------------ |
|
The first thing you should notice is that the QM/MM result has an extra field (PM3ESCF). This is the energy due to the quantum part of the calculation (the NMA) within the presence of the charge field of the MM part (the water). You should also notice here that the QM/MM result lacks ANGLE, DIHEDRAL, 1-4 NB and 1-4 EEL energies. This is exactly as we expect since the NMA bonds, angles, dihedrals and VDW and electrostatic terms are now dealt with inside the QM calculation. The only atoms remaining in the MM section are TIP3P water molecules. These are actually triangulated water and so only have bond terms (TIP3P water does not have an angle component). The water molecules are also only 3 atoms in size and so do not have any 1-4 NB or 1-4 EEL interactions. If you compare the two output files further you should find that the temperatures are reasonably stable and similar. They will not be exactly the same since we are tracking different trajectories. |
Now, lets compare the actual dynamics. First off lets load the QM/MM mdcrd file into vmd:
Fire up VMD and load the NMA topology file (remember to select parm7 [vmd1.8.3] or AMBER7 Parm [vmd1.8.4]) and then load the md_qmmm.mdcrd file into this molecule. Remember, we have used periodic boundaries here so select "crdbox".
We should again remove the water molecules so our NMA is easier to see.
Graphics->Representations
In Selected Atoms type:
all not water
And hit apply
At the same time change the drawing method to CPK.
You should now be able to watch a movie of our NMA molecule "wiggling" about. How does the dynamics of the NMA differ from the classical simulation? If you want you can load both molecules into VMD and animate them together.
Rewind the current QM/MM loaded trajectory to the beginning and then load a new molecule. Select the NMA.prmtop again and then load the md_classical.mdcrd file into this molecule. When you close the load molecule dialog box you should find that both molecules are displayed. You can now play through the trajectory slowly and watch the difference. (You can colour the two molecules differently in Graphics->Representations if you want).
You should notice that the dynamics of the NH group are different in the QM/MM calculation. The nitrogen regularly forms a pyramidal structure and the amine hydrogen is more often out of the O-C-N plane. The QM/MM behaviour here is believed to be the more accurate. The NH backbone dynamics are known to be troublesome to model accurately with classical force fields.
|
|
|
| Classical (MM) | QM |
I leave it to you the reader to try other forms of analysis, you could look at the difference between average structures, plot the O-C-N-H out of plane angle as a function of time. You could also try cluster analysis and hydrogen bond analysis. How does the frequency of hydrogen bonds differ between the classical and the QM/MM simulation? See tutorial 8 for details on this type of advanced analysis.
You could also try coupling this QM/MM approach with Nudged Elastic Band. A classical (MM) introduction to this is given in tutorial 7. By utilizing QM/MM it becomes possible to look at reaction pathways instead of just conformational changes.
| Return to index |
(Note: These tutorials are meant to provide
illustrative examples of how to use the AMBER software suite to carry out
simulations that can be run on a simple workstation in a reasonable period of
time. They do not necessarily provide the optimal choice of parameters or
methods for the particular application area.)
Copyright Ross Walker 2004