Last Updated 28/10/2015
#include <disclaimer.h>
void main()
{
printf ("You use all of my advice and software at your own risk. Enough said...\n");
}
|
The following pages illustrate (and make available for download) several pieces of software that I have written during the course of my research. Most of my time is now spent developing the AMBER suite of Molecular Dynamics Software. Information about this software is available from the main AMBER website: http://amber.scripps.edu Please note: This software carries no warranty whatsoever and has only sketchy documentation. Most of this software was developed for my own use during my research and thus has only limited error trapping. You use it at your own risk. Comments and suggestions are, however, always welcome. |
| GPU Optimized Hardware | AMBER Patches |
| WUWDWA (Unix) | Computational
Chemistry tools (Win9x) | Unique Sequence Identifier |
1) GPU Optimized Life Science and Machine Learning Computing Solutions
In collaboration with Exxact Corp I have designed a series of GPU and Xeon Phi accelerated computing solutions for life sciences and machine learning. This includes the AMBER Certified MD Workstation program, the Gromacs Certified MD Workstation program, the Life Sciences Certified GPU Computing Solutions program, The Intel Xeon Phi Certified Life Sciences Computing program and the Digits and Deep Learning Dev Box program. This provides a simple way to purchase machines that come with Life Sciences or Deep Machine Learning / Convolution Neural Network software preinstalled. The systems come pre-installed and fully tested and are ready to run out of the box. A range of optimized specifications are available for different budget levels and all are fully customizable and include a full warranty. Free test drives of these machines are also available.
2) Various Patches and Utilities for the AMBER Molecular Dynamics Package
For information on the AMBER MD package please see the AMBER website (http://amber.ch.ic.ac.uk/)
1.1) Carnal transparent Bzip2 Support
The following patches add bzip2 decompression support to the reading of coordinate files in the carnal analysis package included within AMBER. You will need to have Bzip2 installed on your system in order for this to work.
Note: Carnal supports gzip and Z-Lib by default.
Programs: Carnal
Description: Most versions of Linux now include support for the bzip2 method of file compression. This typically offers between 10 and 20% higher compression ratios than gzip. This patch adds support for bzipped mdcrd files in carnal using the bunzip2 program. Bzip2 files should have the file extension .bz2
Usage: Apply the following patch to amber6/src/carnal/util.c:
| Downloads | |
|
AMBER 6 Carnal Bzip2 Patch |
AMBER 7 Carnal Bzip2 Patch |
3) What's Up What's Down Who's Around (WUWDWA) [Linux]
This program consists of a series of scripts that are run as part of a cronjob (e.g every 5 minutes) on a linux machine. They monitor a predefined list of IP addresses showing which ones are currently responding and which aren't and then publishes this in the form of a self refreshing web page.
There are currently 3 versions available for download.
1) WUWDWA Version 1.0 (Nov 2000)
This version simply monitors which machines are up by pinging them and then creates a simple web page as shown here.
2) WUWDWA Version 2.1 (Dec 2000)
This version is similar to version 1 but produces a tidier status webpage and also includes rpc.rstatd support by using rup to list both the uptime and load average of machines which support this function. An example webpage is available here.
3) WUWDWA Version 3.0 (Mar 2001)
This version is similar to version 2 but now makes use of a configuration file that allows the path to the wuwdwa.html file to be easily set and for the number of ping retries before a machine is deemed to be offline to be adjusted. There is also a new facility which allows titles to be placed in the machine list file allowing the machine listing to be grouped into categories. An example webpage is available here.
| Downloads | ||
|
WUWDWA v1.0 WUWDWA-v1.0.tar.gz |
WUWDWA v2.1 WUWDWA-v2.1.tar.gz |
WUWDWA v3.0 WUWDWA-v3.0.tar.gz |
Note: These programs require the bash shell in order to work. Also note that these scripts were developed for Linux and so may or may not work on other flavours of unix.
4) Computational Chemistry Tools v1.8.3 [Win9x / NT / 2000 / XP]
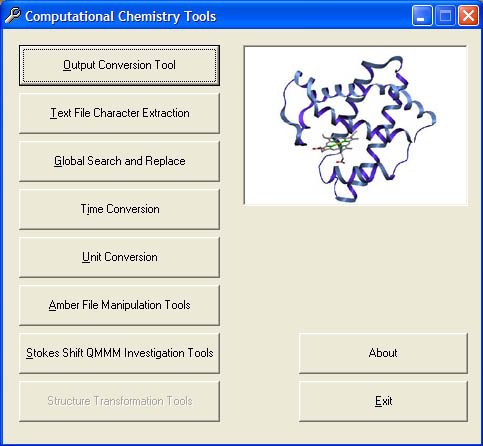
Screenshot of Main Menu - Version 1.4.2
This program is a collection of tools, useful to a computational chemist, that I have developed for my own use during my research, if you want the source code please email me. The modules currently available are:
1) Output Conversion
Converts to and from a number of file formats - specifically it can convert Gaussian 98 Output, Gaussian 94 Output and Cartesian Gaussian Input files to either PDB or XYZ. It can also convert XYZ to PDB and can convert Gaussian 98 Output Files to Gaussian 94 Format.
It can also extract multiple geometries from Gaussian 98 and Gaussian 94 Files to sequentially numbered PDB or XYZ files so that movies can be generated showing exactly how the structure changed during an optimisation.
Screenshot (v1.4.2)
2) Text File Character Extraction.
Provides a windows based solution for extracting columns from text files such as protein databank files or XYZ coordinate files.
Screenshot (v0.9.1)
3) Global Search and Replace
This offers the facility to carry out global text search and replace operations across thousands of files without the need to have all the files currently loaded in memory. The facility to backup the original files before replacement is also provided.
Screenshot (v0.9.1)
4) Time Conversion
This module searches output files from programs such as Gaussian 98 and looks for time strings in the form VV days XX hours YY minutes ZZ.Z seconds and converts it to a plottable format in the units selected. It will also take as input several thousand files and create a single summary file reporting the time line from each file. Typical processing speeds are approximately 50 Mb of text per minute. e.g. 500 x 100 kB files in less than a minute. This will be further improved in later versions.
Screenshot (v0.9.1)
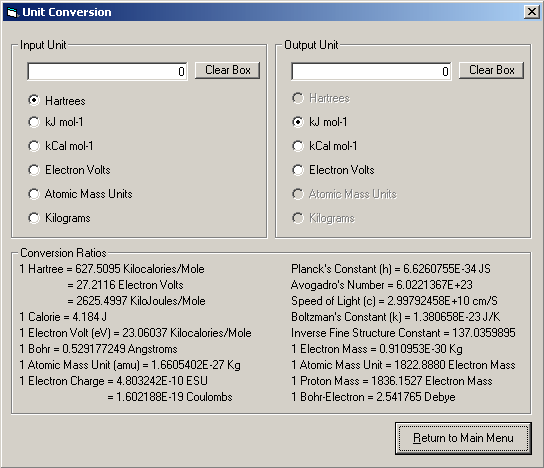
6) Unit Conversion
This module offers a quick and easy conversion between a range of commonly encountered units and also lists typical conversion factors.
Screenshot (v0.9.1)
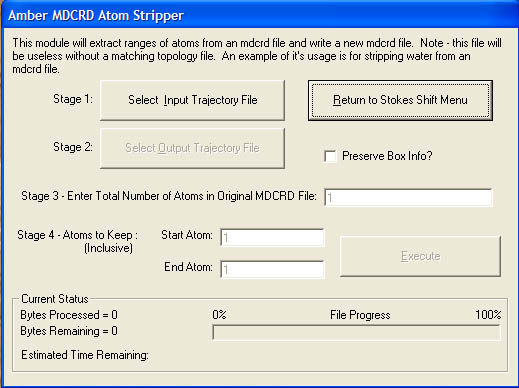
7) Amber File Manipulation Tools
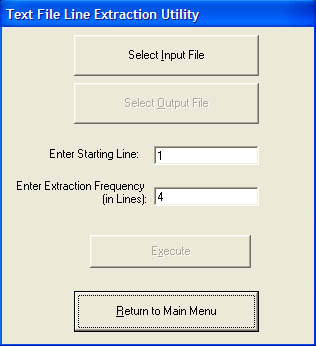
This module is currently development and may not work exactly as expected (you have been warned). Currently it provides two sub modules. The first is for stripping atoms out of Amber trajectory files (this currently does not work with Amber 7). This was developed for modifying trajectory files before opening in VMD, for example stripping the solvent waters. This is necessary because VMD does not work properly for large systems of around 70,000 atoms. Stripping the solvent waters from the trajectory solves the problems.
The second module provides the ability to extract alternate lines from a file. E.g if you have a text file containing an energy on every line but you only want every 3rd line this module is what you need.Screenshot (v1.4.2)
8) Stokes Shift QMMM Investigation Tools
This module was developed for a specific in house task and thus is not covered here.
|
DOWNLOAD |
|
Current Version is 1.8.3 Unzip the file and run setup to install.
comptoolv1-8.zip |
5) Unique Amino Acid Sequence Identifier
This is a short program written in C that can identify the shortest unique sequence (or several shortest unique sequences if there are several of the same length) within a chain of amino acids. It expects a data file in Unix ASCII format that contains comma delimited upper case 3 letter codes for each of the amino acids in the sequence. E.g
CYS,HIS,ALA,PRO,CYS,ALN,HIS
Currently it recognises the 20 common amino acids (ALA,ARG,ASN,ASP,CYS,GLN,GLU,GLY,HIS,ILE,LEU,LYS,MET,PHE,PRO,SER,THR,TRP,TYP,VAL).
Note:- The current algorithm employed is very cpu intensive and has not been
optimised since the program was written for an in house job. Currently if you
are searching for unique lengths greater than about 10 amino acids the search
will take a very long time since the number of combinations searched scales at a
maximum of (20^n)*N where N is the length of the chain being searched and n is
the current length of the unique sequence being searched for. Typically,
however, scaling will be well below this maximum. I do not recommend it though
for searching where you expect the shortest unique chain length to be greater
than 10 amino acids. A new, more efficient algorithm will be added at a later
date.
Usage:
./a.out -i sequence.dat -a 1 -s 1
-i = Input data
filename
-a = Search algorithm to use -
currently only method 1 is supported
-s = Start length to begin search at.
E.g -s 5 will only report a unique sequence if it contains 5 or more amino
acids.
|
DOWNLOAD |
|
Current Version is 0.1.2 Unzip the file and compile with a suitable c compiler.
Unique_sequence_id_v0.1.2.zip |